
Your Source for Cutting-Edge Research
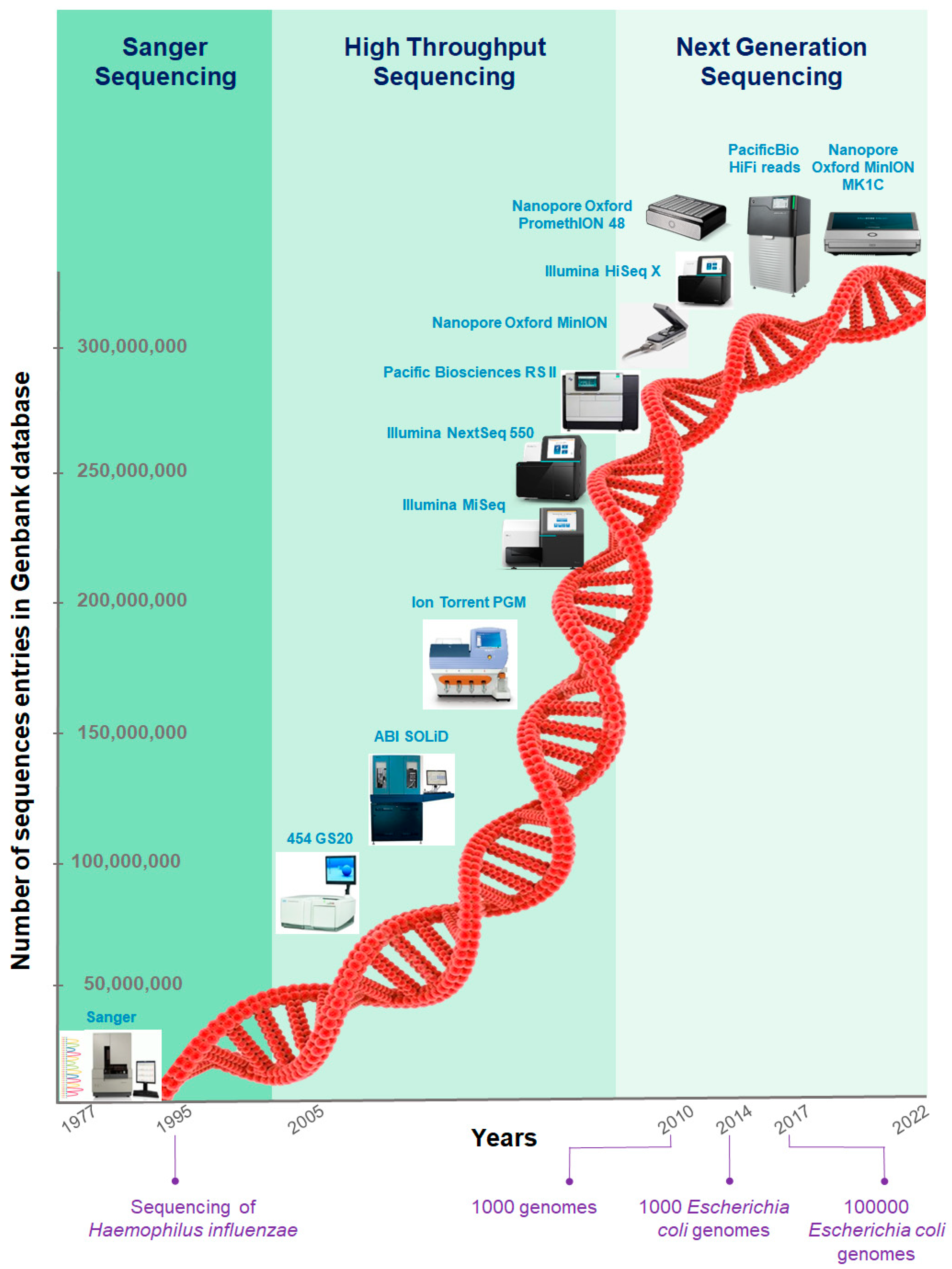
For organisms with no reference genome or highly dynamic genomes, DNA-sequencing data analysis starts with assembling a genome de novo. Genome assembly benefits from deep whole-genome sequencing.
An assembled genome is annotated based on sequence homology, predicted gene sequences and, if available, RNA-sequencing data from the same organism. If annotated genomes for close relative species exist, the annotation can be improved by transferring gene information to the newly assembled genome.
The quality of an assembled genome is assessed using metrics such as N50, L50, and completeness with regards to highly conserved orthologs. A new high-quality genome enables analyses into pan-genomes, population genetics, and much more.
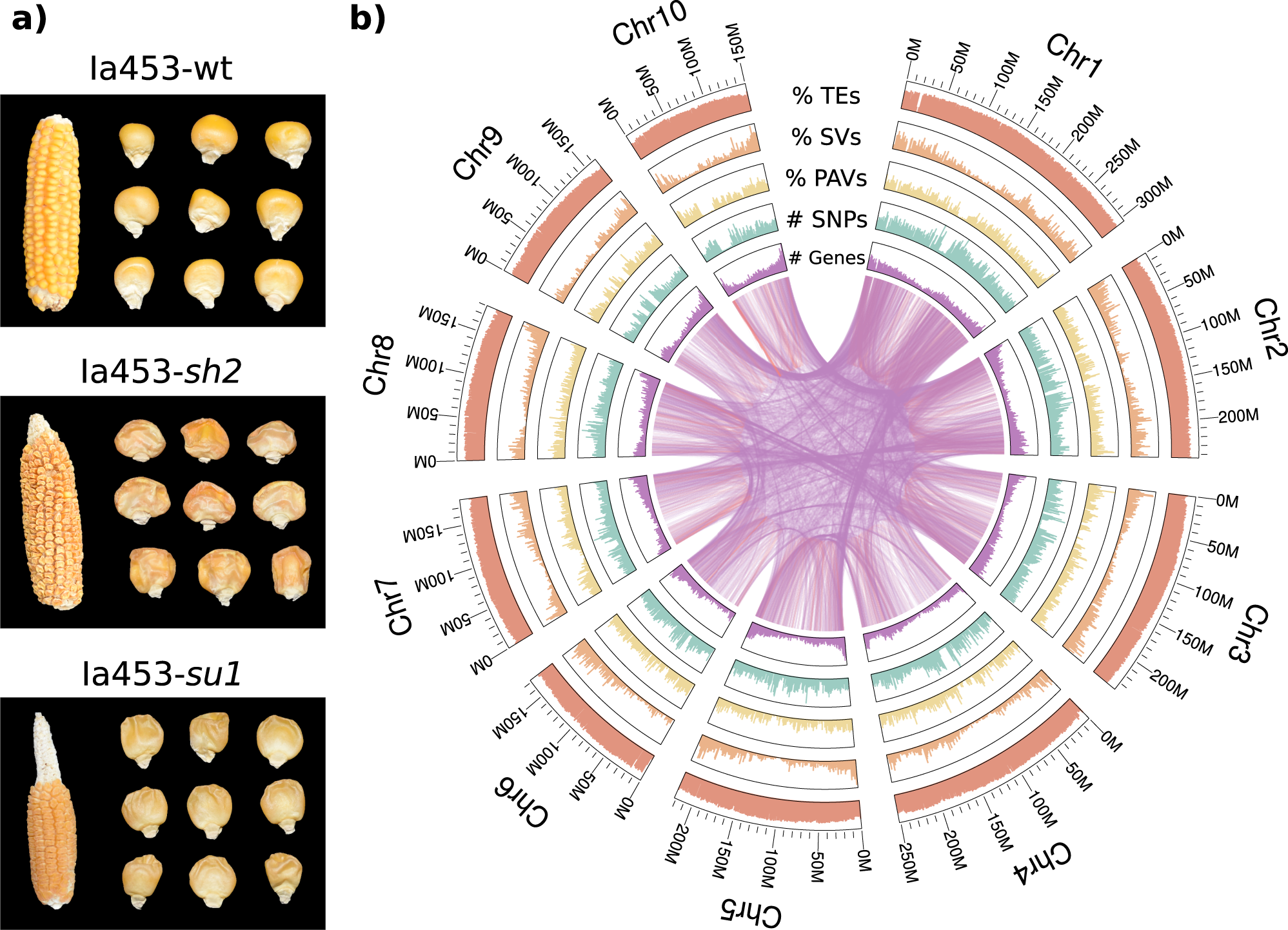
Genome-Wide measurement of individuals sampled from related populations contain rich information on the populations’ structure, genealogy, and history. Population genetic analyses of non-model organisms often begin with genome assembly and annotation, and proceed to identifying genetic polymorphisms in the sampled populations. The downstream analyses based on these polymorphisms and their allele frequencies help study evolutionary phenomena such as speciation and adaptation.
Typical analyses involve principal component analysis, analysis of genetic variation within and between populations to identify loci affected by evolutionary selection, and analyses of population admixtures, phylogeny, and demographic histories.
biomedically motivated population-scale genetic analysis aim to identify genes and variants associated with relevant phenotypes or diseases. Apart from the few diseases which are monogenic and strongly heritable, most diseases require large, population-level sample sizes to achieve sufficient statistical power to find associations. Such genome-wide association studies (GWAS) are based on SNP-array or DNA-sequencing data from biobanks or other large repositories.
GWAS results in summary statistics on the association between each individual variant and the studied disease. In the case of polygenic diseases, individual variants may have very weak effect sizes even when the disease is strongly heritable. In such cases, polygenic risk scores (PRS) can be used to sum the effect of a large number of variants, resulting in a combined risk score with potential clinical utility.